Overview
ChemMORT (Chemical Molecular Optimization, Representation and Translation) is a platform which provides chemical space navigation with possibilities for de novo molecular optimization. Based on the combination of Neural Machine Translation model and Particle Swarm Optimization method, ChemMORT is able to accomplish multi-parameter optimization tasks effectively. Three modules are provided: SMILES Encoder, Descriptor Decoder and Molecular Optimizer.
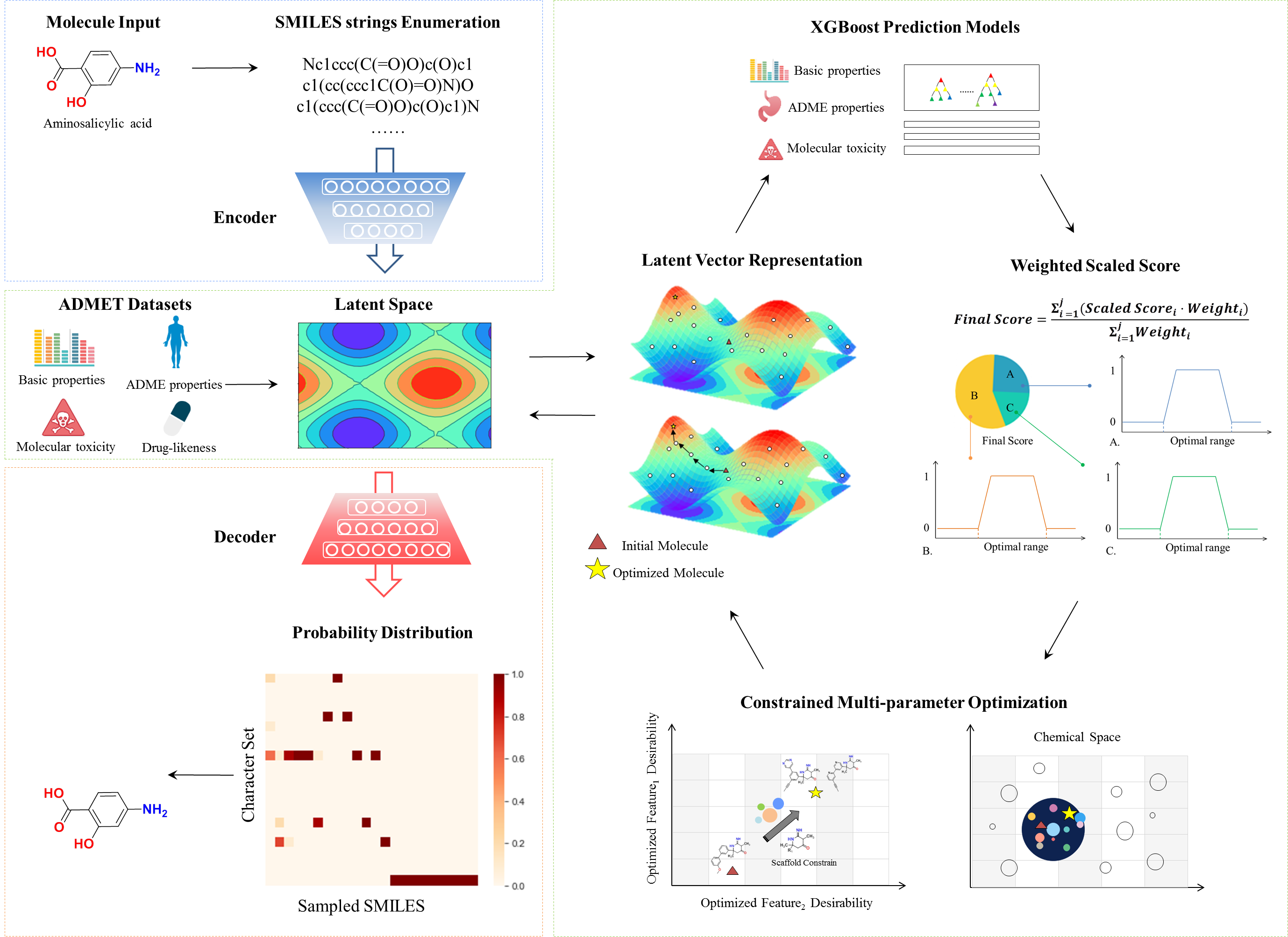
Figure 1. Diagram of workflow
Introduction
The SMILES Encoder allows the user to transform the SMILES string to a 512-dimensional vector through the application of well-trained encoder network from Neural Machine Translation model. Owing to its consecutive, reversible and informative characters, such representation is recommended for the use of a sort of molecular descriptors or GPS for the chemical space of the molecules. It provides four types of inputs to start encoding: single/batch SMILES string(s) input, file upload (*.sdf/*.csv/*.txt)and molecule drawing from editor.
The Embedding Decoder is able to back-engineer the 512-dimensional vector to the corresponding uniform canonical SMILES string through the application of well-trained decoder network from Neural Machine Translation model. Such function makes it possible to be a steered solution for molecular de novo optimization, as the possibility output of the decoder can be sampled. User can upload file (*.csv) for decoding, which provide information about 512-dimensional vector characterization.
The Molecular Optimizer, combining the Neural Machine Translation model and Particle Swarm Optimization method, is designed to optimize molecular features through the application of credible ADMET prediction models. As molecular optimization requires the balance of several criteria, Molecular Optimizer allows the multi-parameter optimization within customized constraints. 11 high-quality ADMET prediction models based on the calculated 512-dimensional vectors are provided in the module (details see Property of Optimizer), which enables the accurate generation of the optimized molecules.
|
Endpoint |
Description |
Performance |
Type |
Method |
|
logD7.4 |
Log of the octanol/water distribution coefficient at pH7.4.
|
Test Set
|
Basic property |
XGBoost |
|
AMES |
The probability to be positive in Ames test.
|
Test Set
|
Toxicity |
XGBoost |
|
Caco-2 |
Papp (Caco-2 Permeability)
|
Test Set
|
Absorption |
XGBoost & Data Augment |
|
MDCK |
Papp (MDCK Permeability) |
Test Set
|
Absorption |
XGBoost & Data Augment |
|
PPB |
Plasma Protein Binding
|
Test Set
|
Distribution |
XGBoost |
|
QED |
Quantitative estimate of drug-likeness |
n/a |
Drug-likeness score |
Molecular Function |
|
SlogP |
Log of the octanol/water partition coefficient, based on an atomic contribution model [Crippen 1999].
|
Fitted on an extensive training set of 9920 molecules, with R2 = 0.918 and σ = 0.677 |
Basic property |
Molecular Function |
|
logS |
Log of Solubility
|
Test Set
|
Basic property |
XGBoost |
|
hERG |
The probability to be hERG Blocker
|
Test Set
|
Toxicity |
XGBoost |
|
Hepatoxicity |
The probability of owning liver toxicity
|
Test Set
|
Toxicity |
XGBoost |
|
LD50 |
LD50 of acute toxicity
|
Test Set
|
Toxicity |
XGBoost |
Development Environment
|
Third party library |
Version |
|
TensorFlow |
1.14.0 |
|
Scikit-learn |
0.23.2 |
|
RDKit |
2019.03.1 |
|
Django |
2.2 |
|
XGBoost |
1.2.0 |
|
Celery |
4.4.7 |
|
RabbitMQ |
3.6.10 |
SMILES Encoder
Three input types are provided for SMILES encoding:
- 1. By inputting SMILES strings
- 2. By uploading file (*.sdf/*.csv/*.txt)
- 3. By drawing molecule from editor below
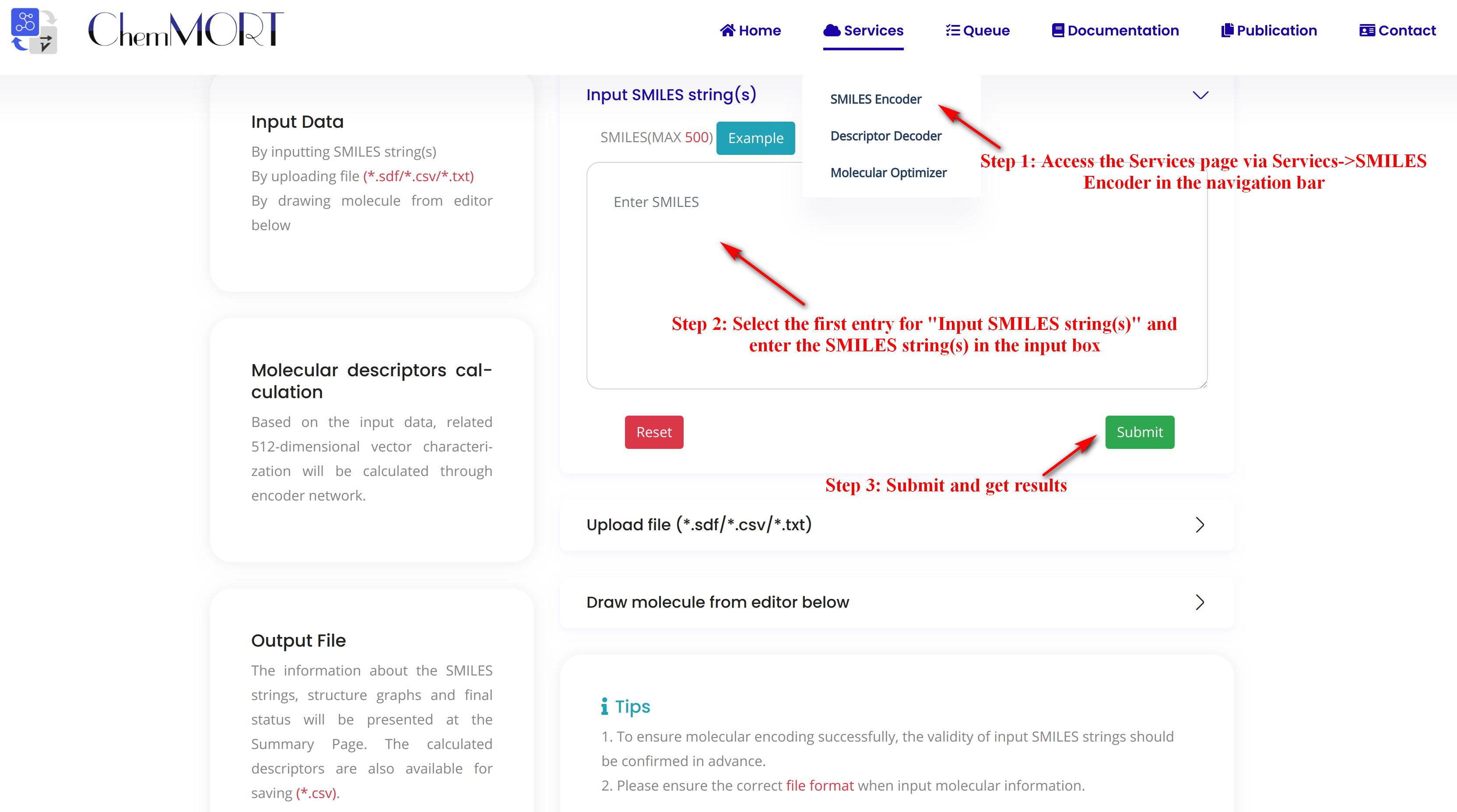
Step1: Access the Services page via Services->SMILES Encoder in the navigation bar.
Step2: Select the first entry for “Input SMILES string(s)” and enter the SMILES string(s) in the input box.
Click the EXAMPLE button to quickly fill in the example SMILES.
Step3: Submit and get results.
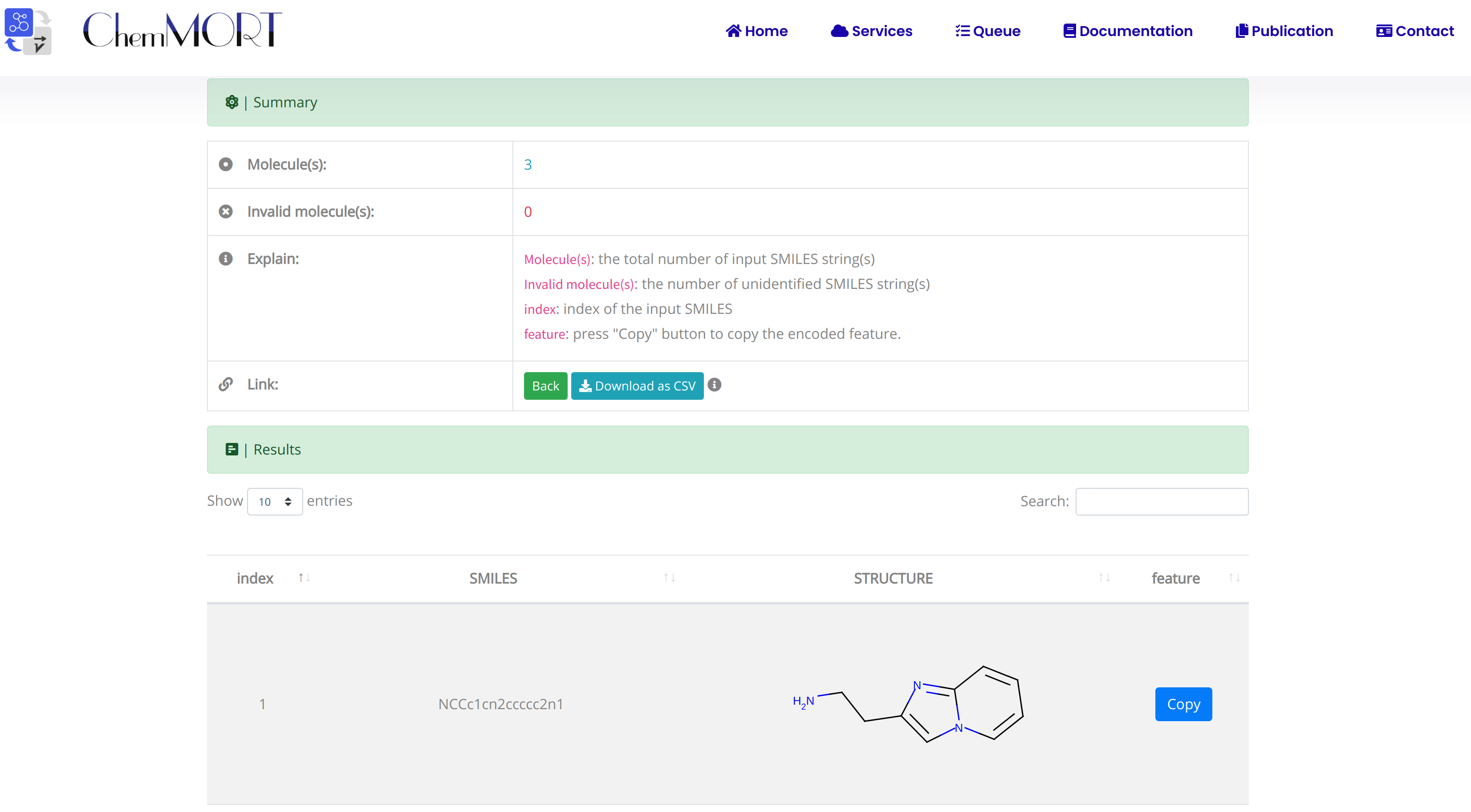
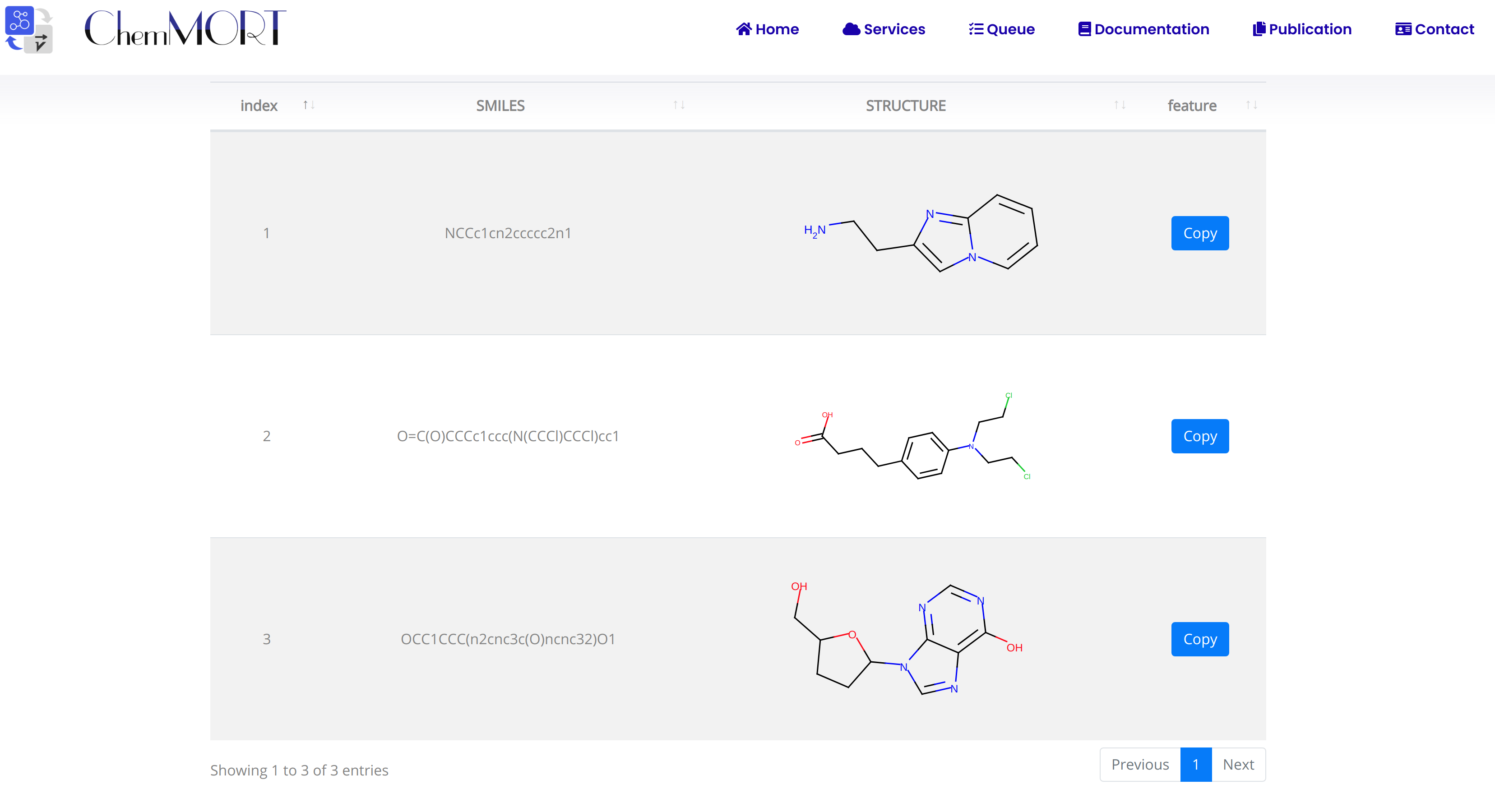
After submission, the input SMILES strings will be transformed to the corresponding 512-dimensional vector representations through the application of well-trained encoder network. In this page, the Summary and Result block will present the overview of the results and the detailed information about the SMILES strings, structure graphs and final status, respectively.
1. In the Summary block:
- 1) Molecules indicates the total number of input SMILES strings;
- 2) Invalid molecules indicates the number of unidentified SMILES string(s).
2. In the Results block:
- 1) index indicates the index of the input SMILES;
- 2) structure is the molecular image;
- 3) SMILES is the input SMILES string;
- 4) Feature "Copy" button allows the copy of the encoded features.
One type of output file is available for downloading, which provides information including the original SMILES and the encoded successful 512-dimensional vectors.
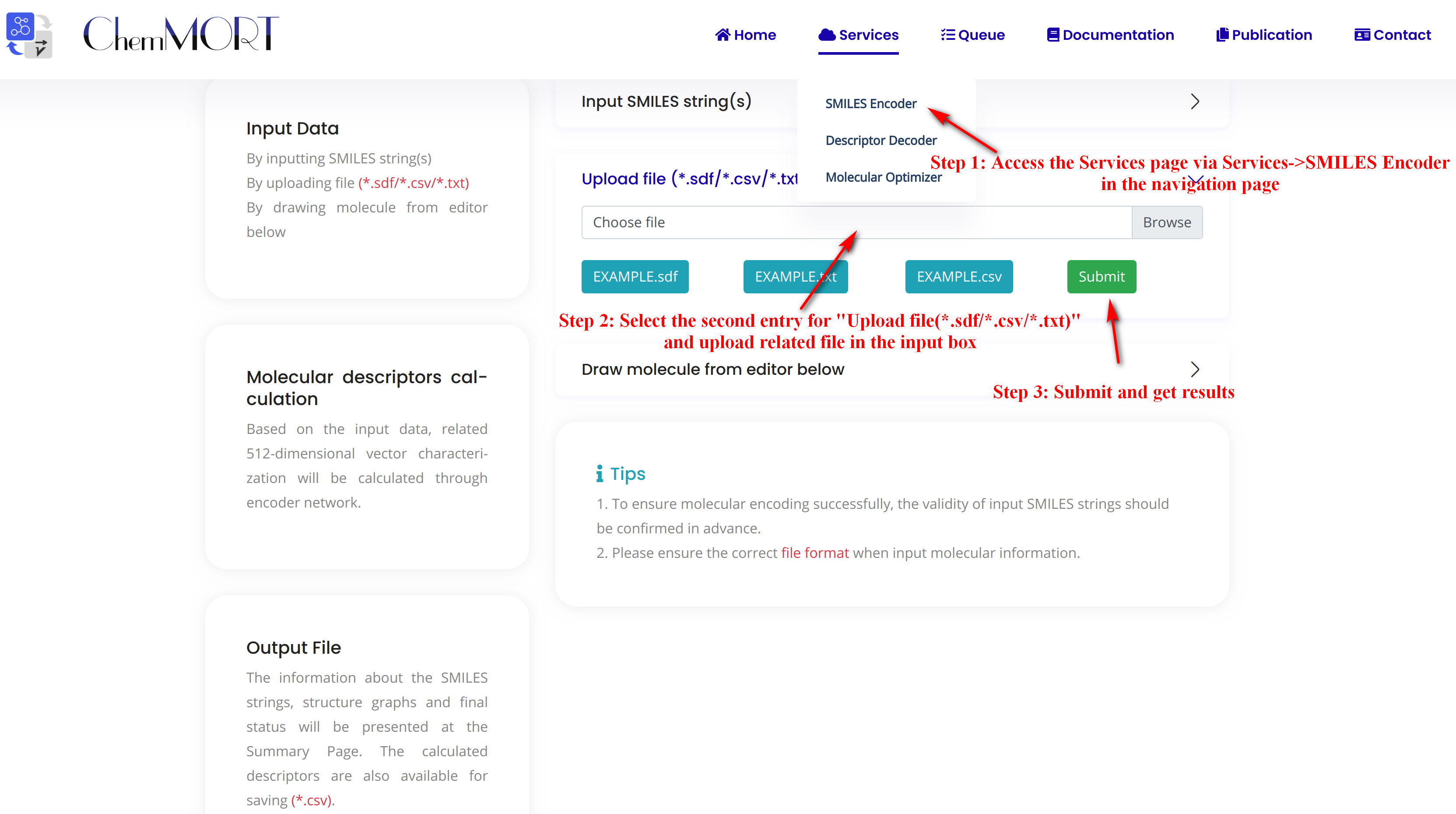
Step1: Access the Services page via Services->SMILES Encoder in the navigation bar.
Step2: Select the second entry for “Upload file (*.sdf/*.csv/*.txt)” and upload related file in the input box.
There are three types of files available for upload here, all of which are in a similar format (*.TXT, *.CSV, *.TSV).
Step3: Submit and get results.
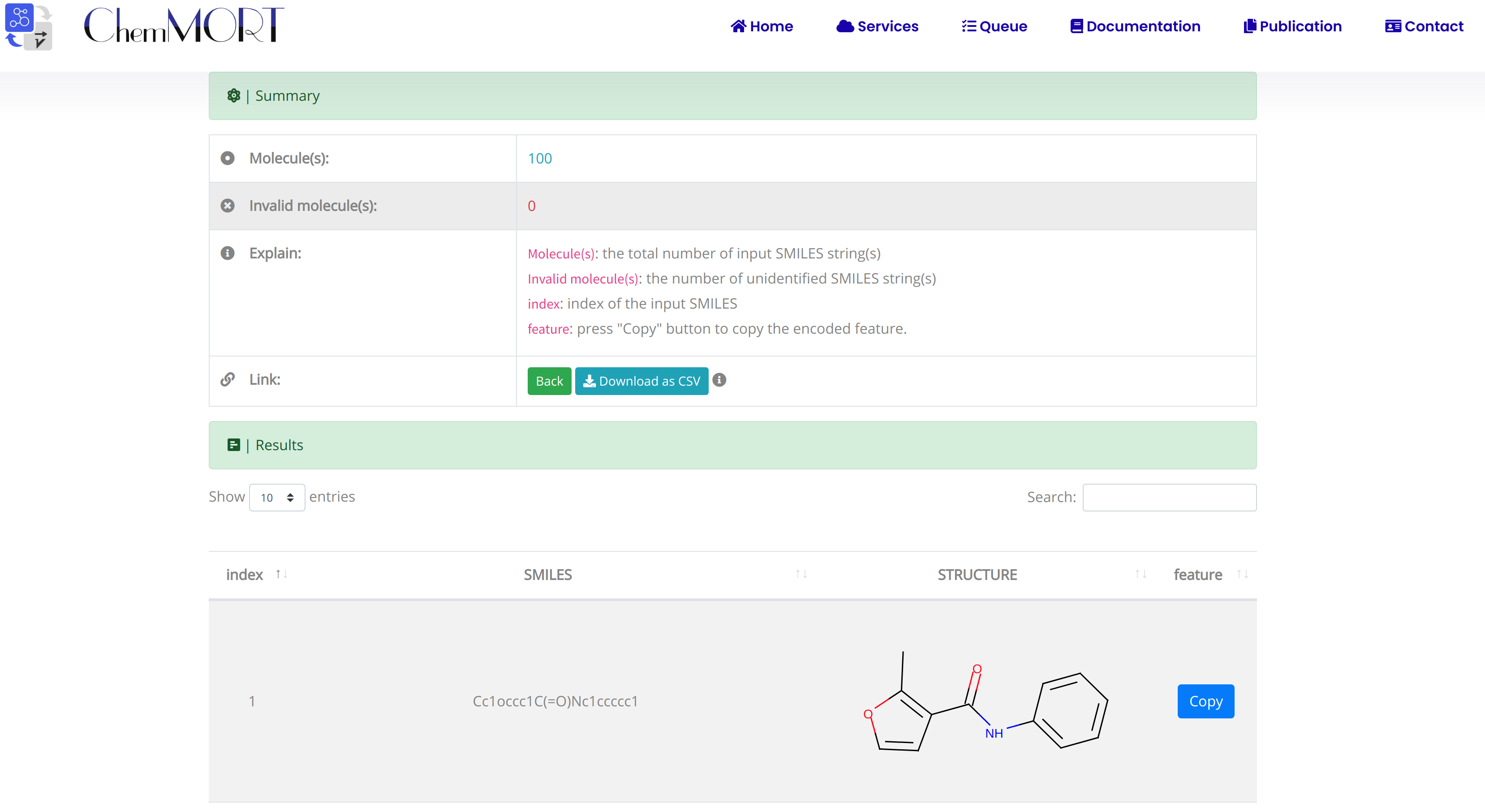
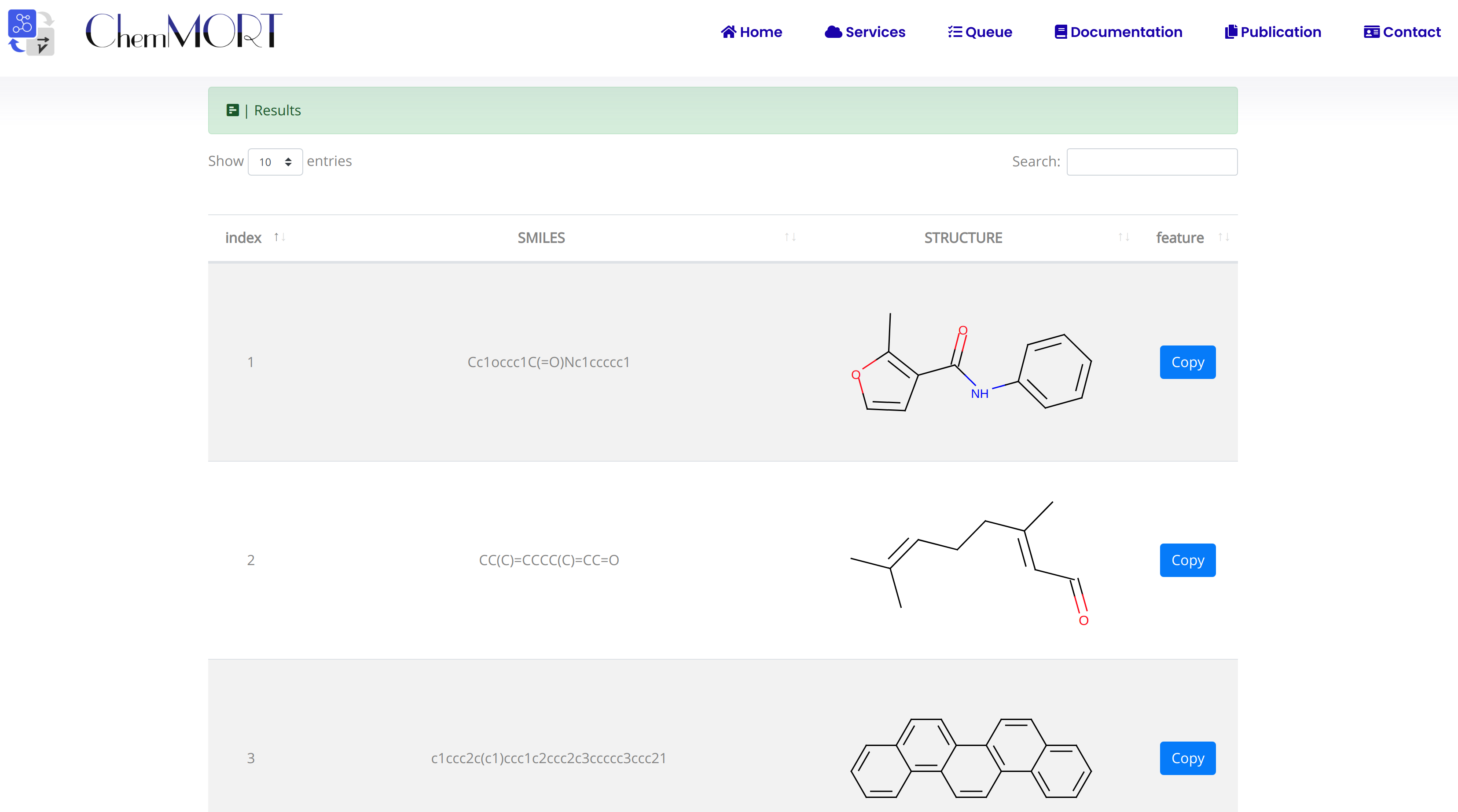
After submission, the input SMILES strings will be transformed to the corresponding 512-dimensional vector representations through the application of well-trained encoder network. In this page, the Summary and Result block will present the overview of the results and the detailed information about the SMILES strings, structure graphs and final status, respectively.
1. In the Summary block:
- 1) Molecules indicates the total number of input SMILES strings;
- 2) Invalid molecules indicates the number of unidentified SMILES string(s).
2. In the Results block:
- 1) index indicates the index of the input SMILES;
- 2) structure is the molecular image;
- 3) SMILES is the input SMILES string;
- 4) Feature "Copy" button allows the copy of the encoded features.
One type of output file is available for downloading, which provides information including the original SMILES and the encoded successful 512-dimensional vectors.
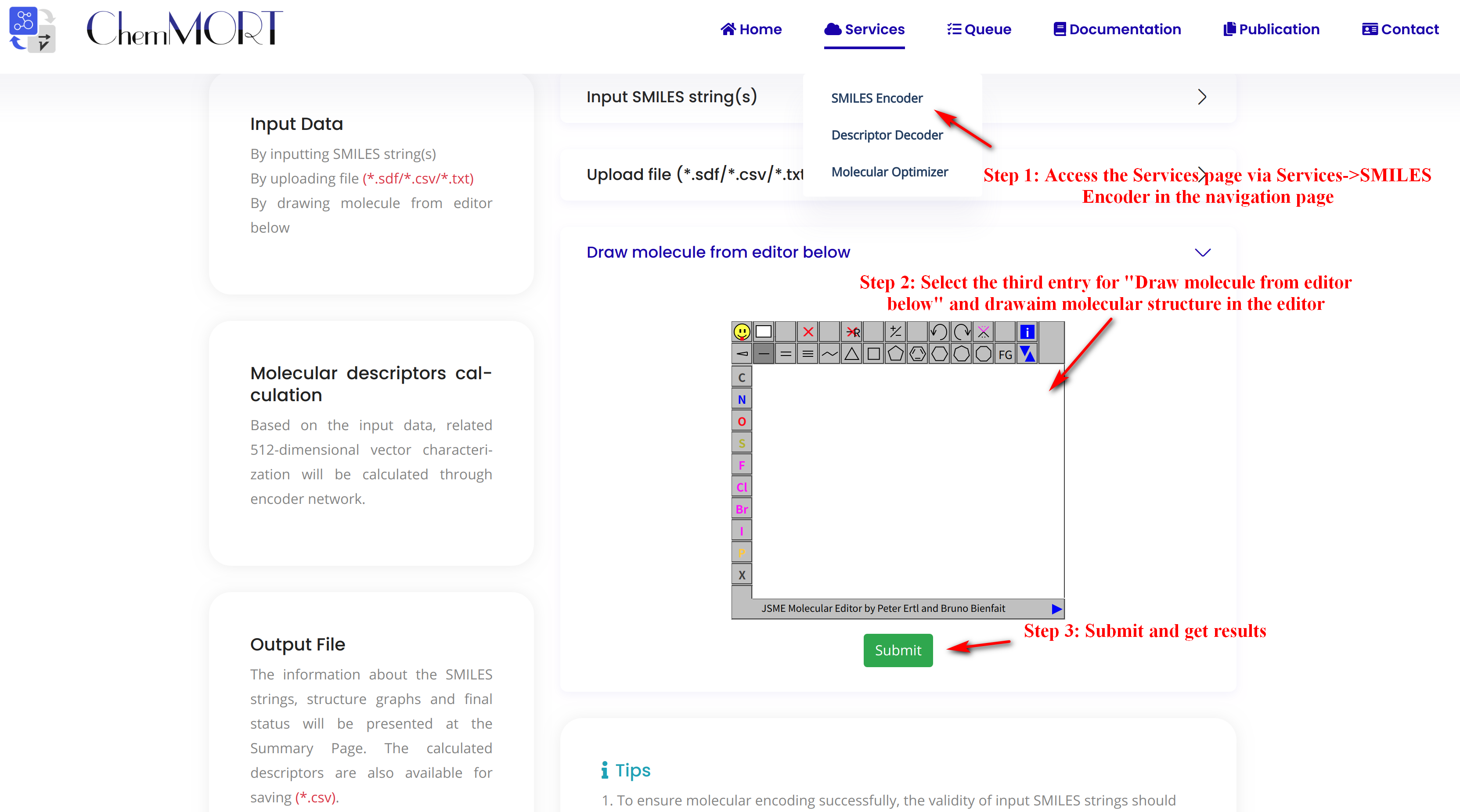
Step1: Access the Services page via Services->SMILES Encoder in the navigation bar.
Step2: Select the third entry for “Draw molecule from editor below” and draw aim molecular structure in the editor.
Step3: Submit and get results.
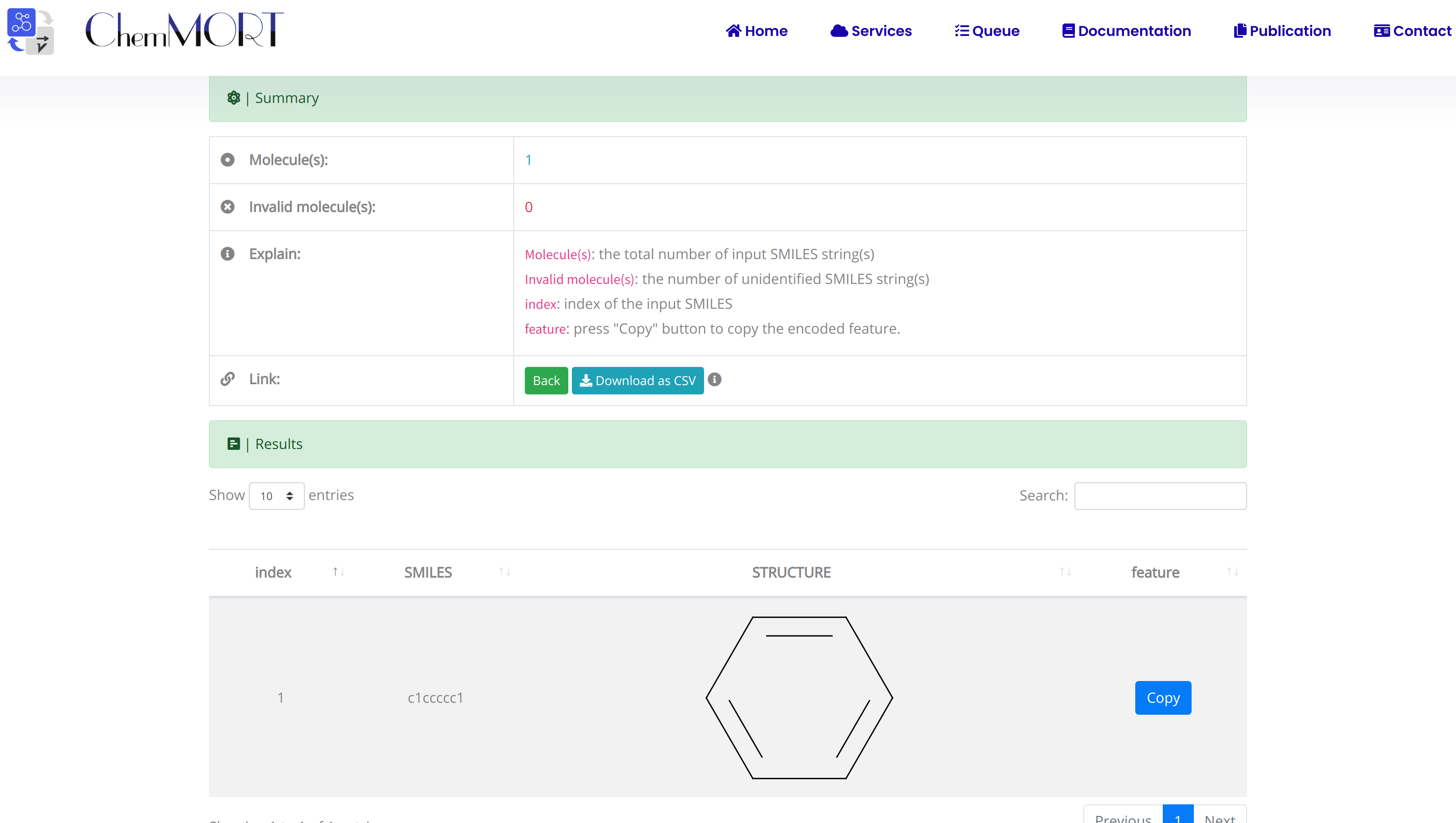
After submission, the input SMILES strings will be transformed to the corresponding 512-dimensional vector representations through the application of well-trained encoder network. In this page, the Summary and Result block will present the overview of the results and the detailed information about the SMILES strings, structure graphs and final status, respectively.
1. In the Summary block:
- 1) Molecules indicates the total number of input SMILES strings;
- 2) Invalid molecules indicates the number of unidentified SMILES string(s).
2. In the Results block:
- 1) index indicates the index of the input SMILES;
- 2) structure is the molecular image;
- 3) SMILES is the input SMILES string;
- 4) Feature "Copy" button allows the copy of the encoded features.
One type of output file is available for downloading, which provides information including the original SMILES and the encoded successful 512-dimensional vectors.
Descriptor Decoder
The Embedding Decoder is able to back-engineer the 512-dimensional vector to the corresponding uniform canonical SMILES string through the application of well-trained decoder network.

Step1: Access the Services page via Services->Descriptor Decoder in the navigation bar.
Step2: Upload file with specified format: one 512-dimensional vector presents per row, where each dimensional value is separated by space. Here's an example:
example.csvStep3: Submit and get results.
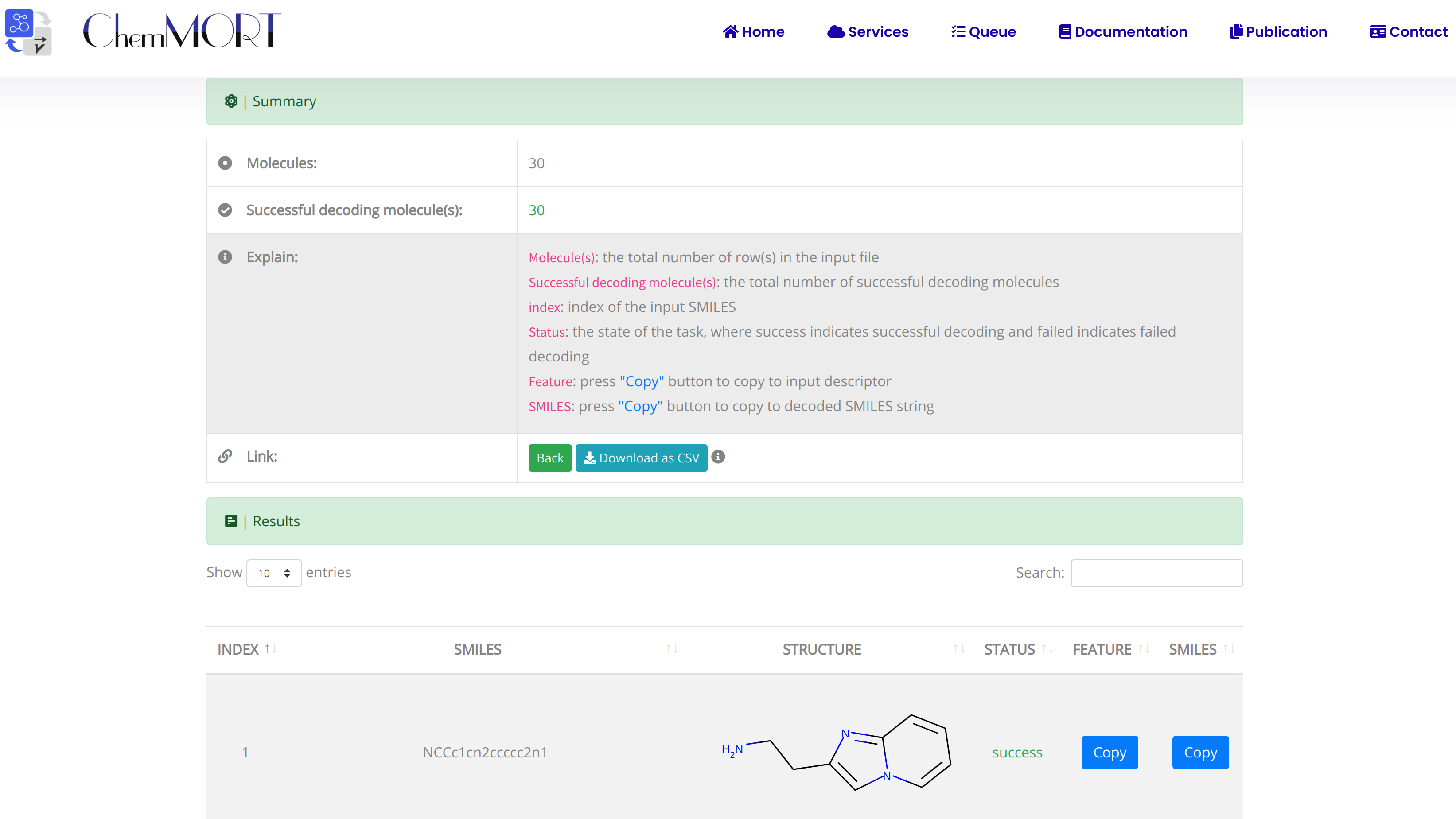
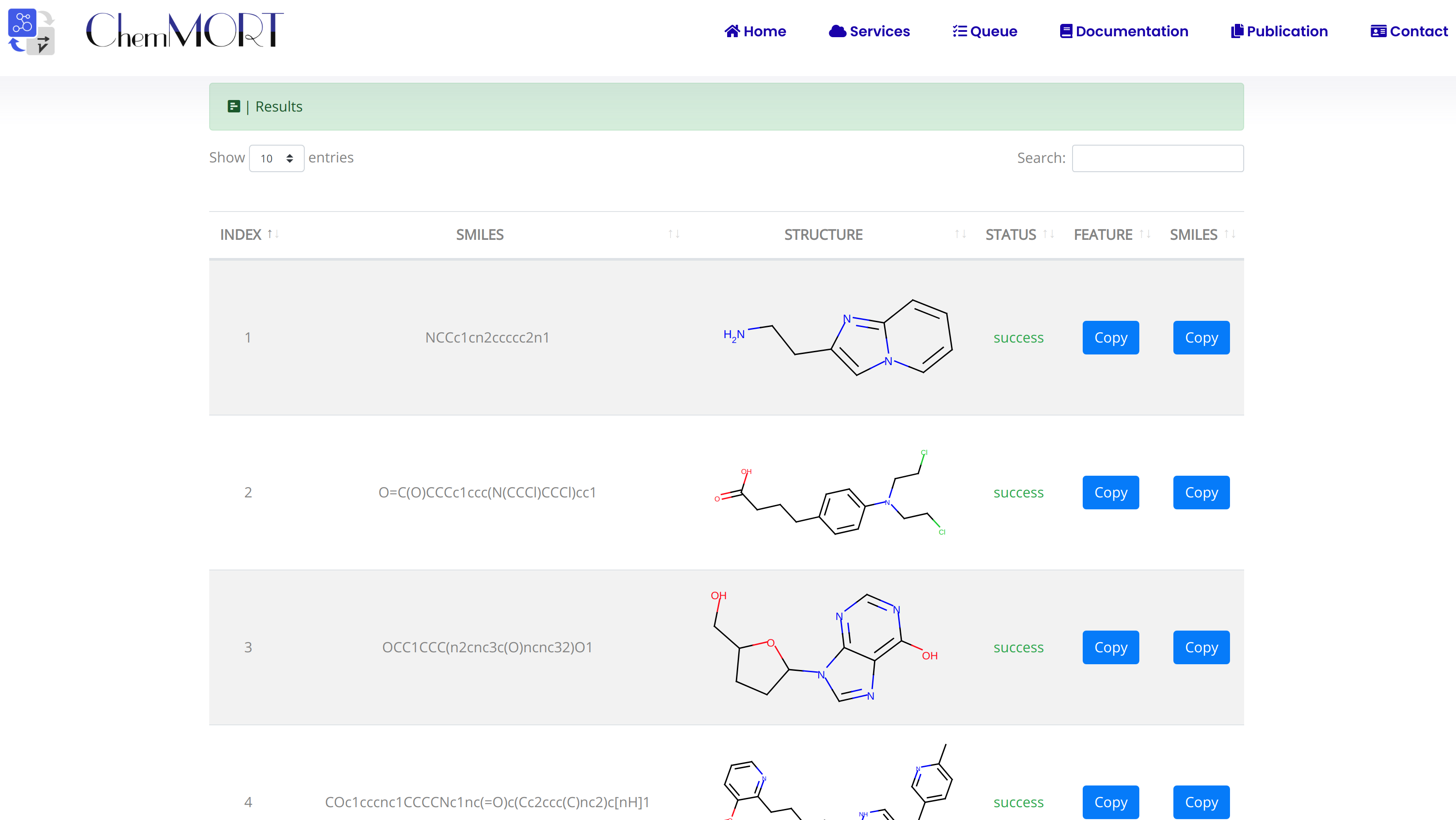
After submission, the input SMILES strings will be transformed to the corresponding 512-dimensional vector representations through the application of well-trained encoder network. In this page, the Summary and Result block will present the overview of the results and the detailed information about the SMILES strings, structure graphs and final status, respectively.
1. In the Summary block:
- 1) Molecules indicates the total number of input SMILES strings;
- 2) Invalid molecules indicates the number of unidentified SMILES string(s).
2. In the Results block:
- 1) index indicates the index of the input SMILES;
- 2) structure is the molecular image;
- 3) SMILES is the input SMILES string;
- 4) Feature "Copy" button allows the copy of the encoded features.
One type of output file is available for downloading, which provides information including the original SMILES, status and the encoded successful 512-dimensional vectors.
Molecular Optimizer
The Molecular Optimizer is designed to optimize molecular features through the application of credible ADMET prediction models. As molecular optimization requires the balance of several criteria, Molecular Optimizer allows the multi-parameter optimization within customized constraints.
Step1: Input the information about job name and email address for the receiving of optimized result;
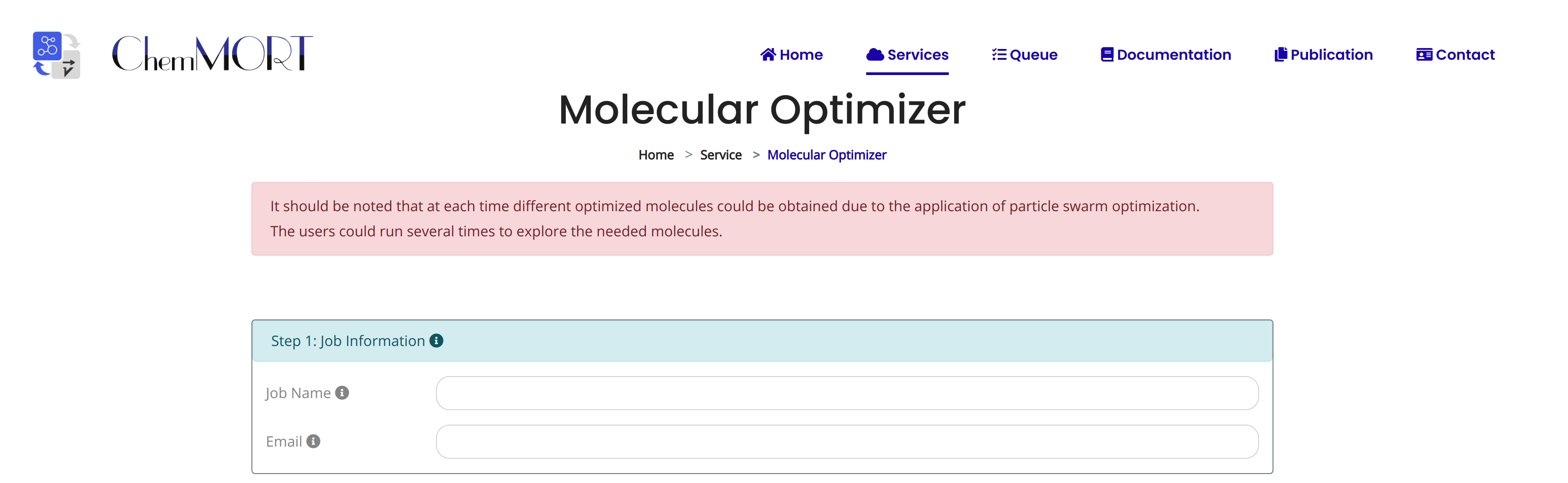
Step2: Input the SMILES string of the target molecule for following optimization;

Click the EXAMPLE button to quickly fill in the example SMILES.
Step3: Select the aim property for optimization with the ideal value or range;
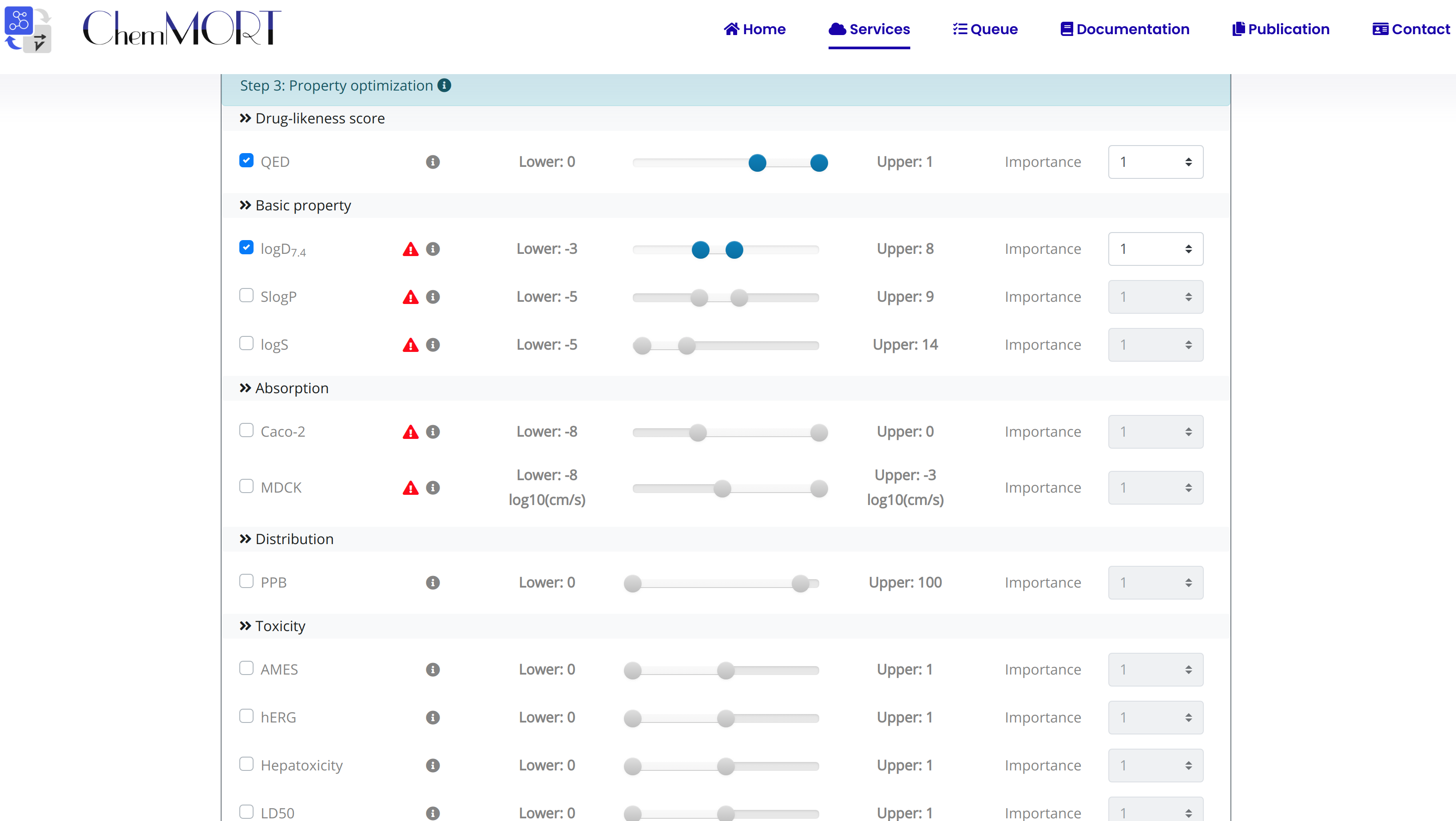
Five feature categories, including basic property, Absorption, Distribution, Drug-likeness score and toxicity, are provided for optimization. To accomplish multi-parameter optimization, several parameters are provided following:
2. Lower and Upper: the scope of the ideal scope of the target property;
3. Score: the index used to evaluate the desirability of the optimized molecule.
` \text {Score}=\frac{\sum_{i=1}^{\j}(\text {Scaled Score}_i \cdot Weight_i)}{\sum_{\i=1}^{\j}Weight_i} `
Where j is the number of aim properties, Scaled Scorei represents the desirability of the aim property i of the optimized molecule, and Weighti represents the contribution of the aim property i of the optimized molecule.
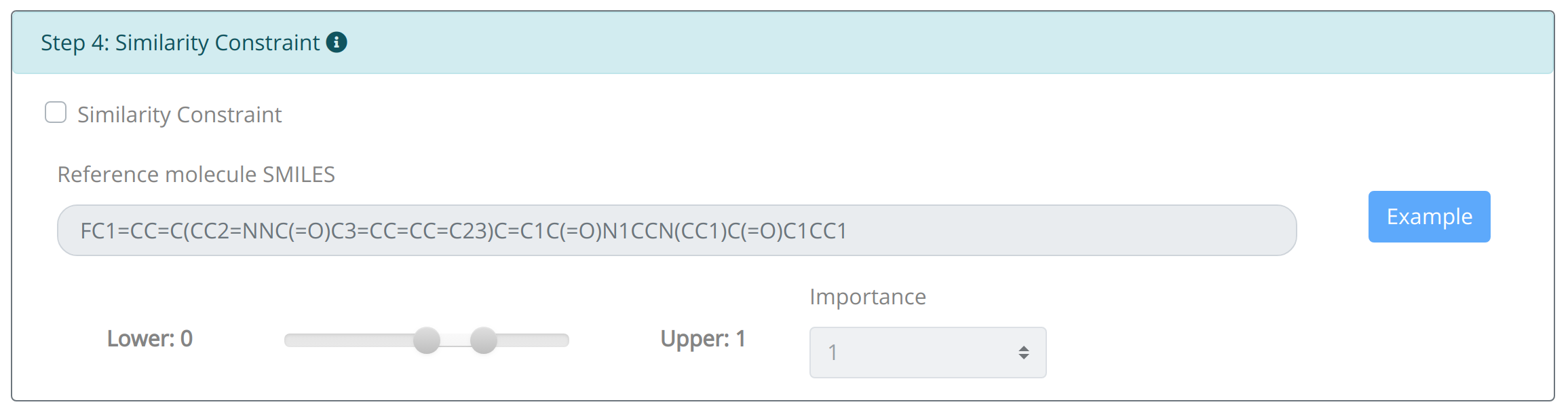
Step4: Set distance constraint:
Though the application of credible optimization objectives can assist the molecular optimization in this endeavor, without any constraints they may solely focus on the very objective thus resulting unpleasant structures. Owing to the nature of structure–activity relationship, Distance Constraint enables the set of distance limitation between generated molecule and the reference molecules based on the application of ECFP4 fingerprint and Tanimoto similarity algorithm. It is also allowed to navigate generated molecule to possess totally different structural features of the initial molecule. The weight set of the distance constraint is similar to the namesake of properties.
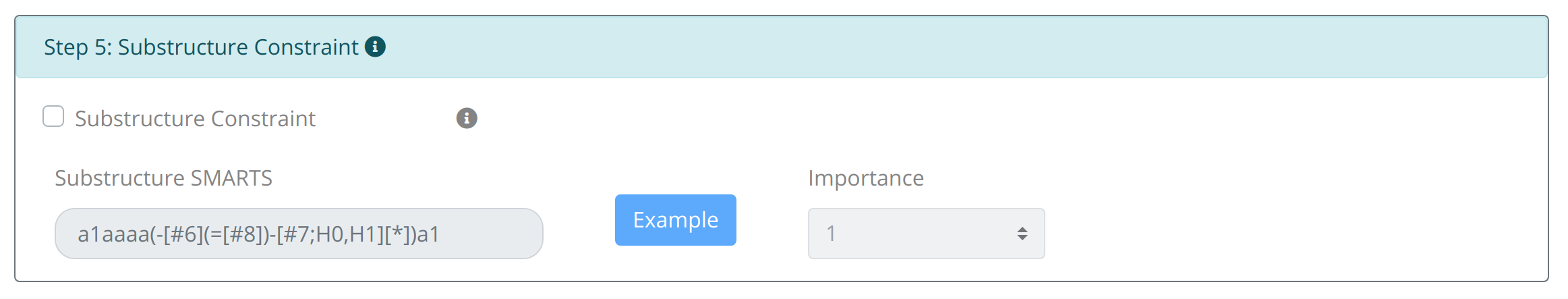
Step5: Set Substructure Constraint:
The constraint about the appearance of important substructure (such as active motif or undesirable substructure) is allowed by inputting corresponding SMARTS in Substructure Constraint. The weight set of the distance constraint is similar to the namesake of properties.
Step6: Set the optimization steps and related circles:
Considering the different needs of calculated time and the number optimal molecules, users can set STEP and TRACK hyperparameters to control the optimization process, where STEPS indicates the number of iterations and TRACK indicates the number of the optimal molecules for displaying.
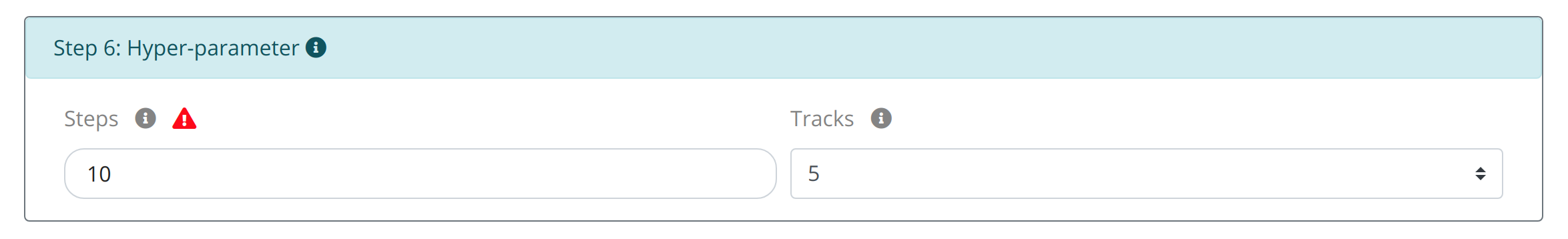
Step7: Submit and get result.
After submission, the task will run automatically in the background. Once it completed, users can view the results via the link provided in the email.
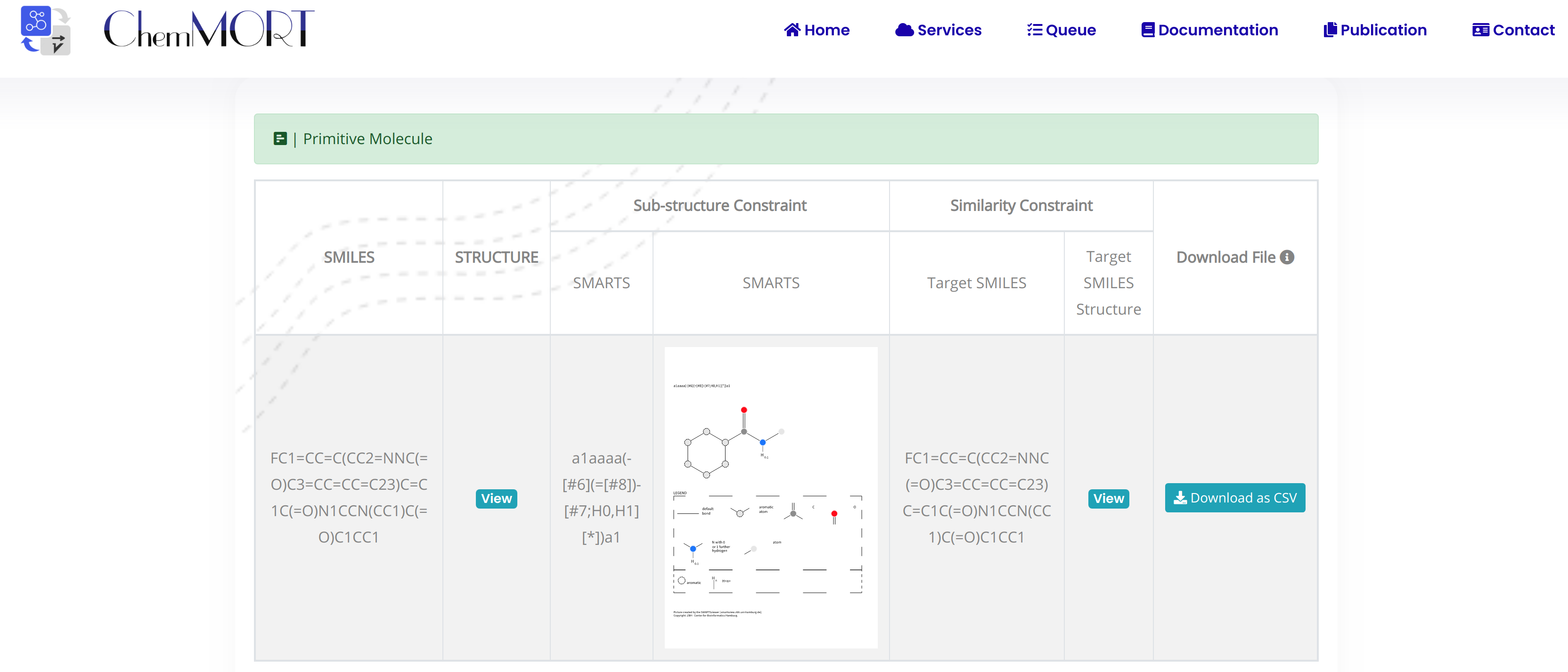
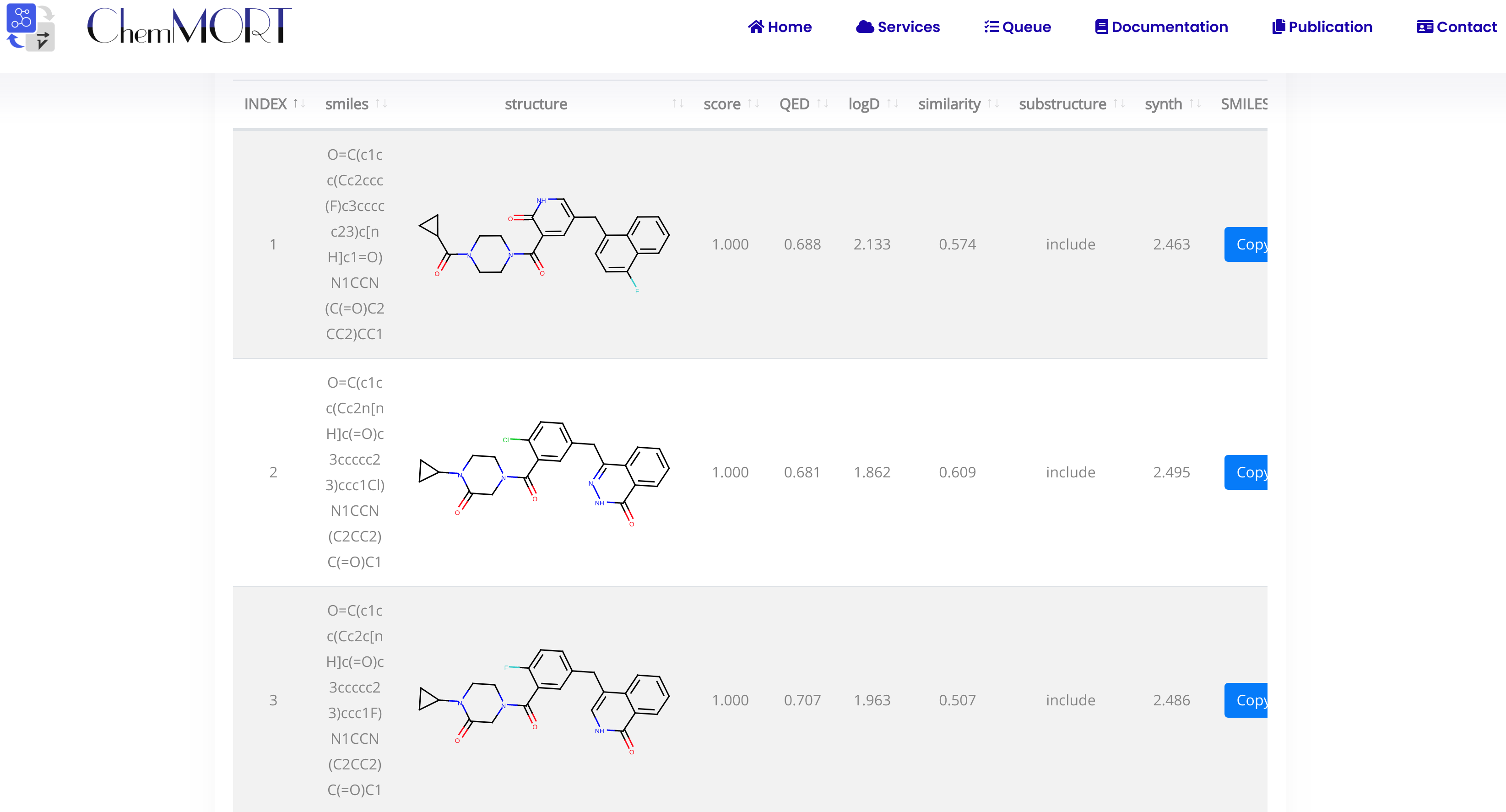
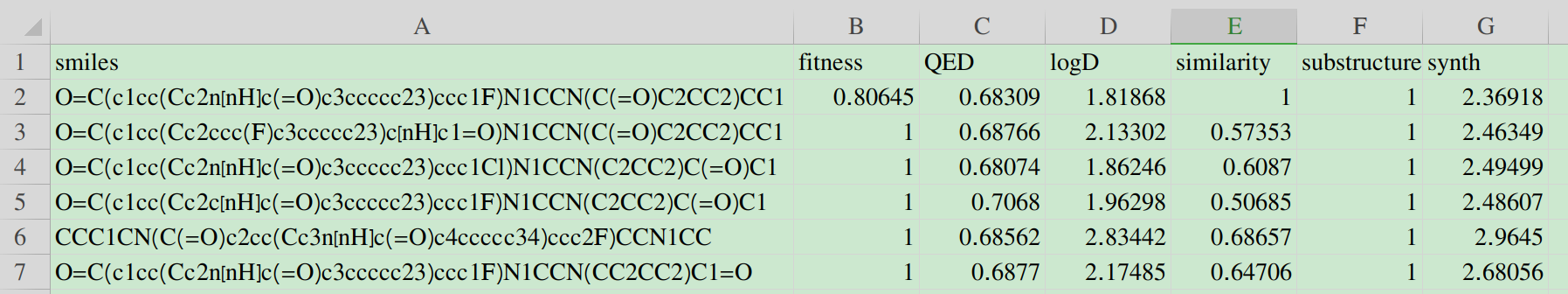
The first row is the information of the initial molecule, and the rest is the information of the optimized molecule.